




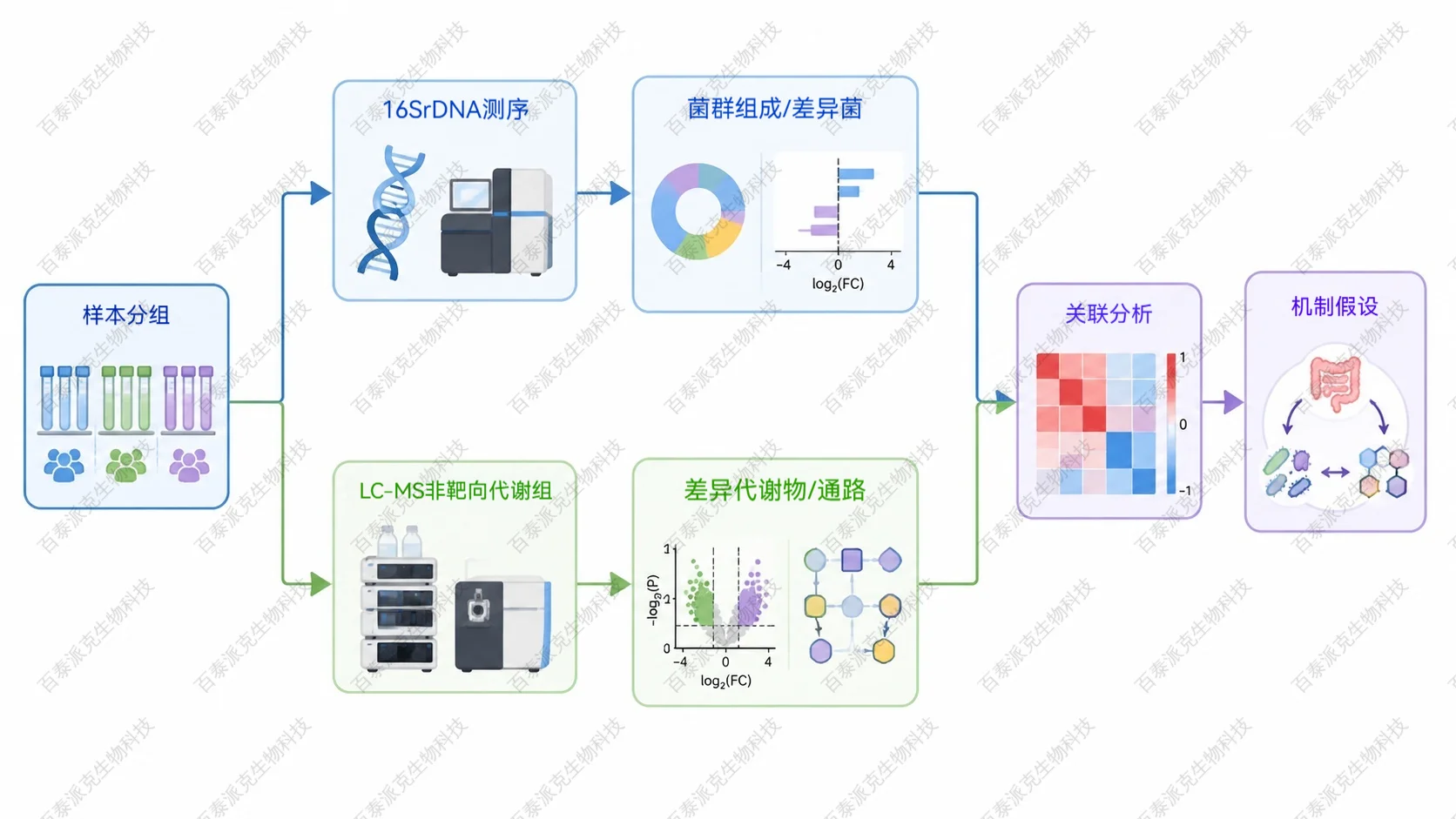
16S 和非靶向代谢组学联合分析:从菌群变化到代谢机制解释
- 16S 多数情况下只能提供分类水平信息,不能直接等同于菌株功能或基因表达。
- 非靶向代谢组学会产生大量特征峰,部分代谢物只能达到推测注释,仍需标准品或靶向方法确认。
- 相关性不等于因果关系,菌群与代谢物之间的变化可能由共同的处理因素驱动。
- 批次效应、样本量不足、分组不平衡和饮食/环境变量都会影响整合分析可靠性。
- 两类数据的尺度、缺失值、归一化方法不同,分析前需要明确质量控制和统计策略。

16S 和非靶向代谢组学联合分析,是把微生物群落结构与小分子代谢物谱放在同一研究框架中解释。它不能只回答“哪些菌变了”或“哪些代谢物变了”,更重要的是帮助研究者判断菌群变化是否与代谢通路、疾病表型、药物干预或环境处理存在一致的关联。
关键要点
|
关键问题 |
简短结论 |
|---|---|
|
联合分析解决什么问题? |
把菌群丰度变化与代谢物变化关联起来,寻找可能参与表型变化的菌-代谢物线索。 |
|
16S 提供什么信息? |
主要提供细菌或古菌群落组成、丰度、α/β 多样性和差异菌群。 |
|
非靶向代谢组提供什么信息? |
提供样品中尽可能广泛的小分子代谢物特征、差异代谢物和通路富集结果。 |
|
结果能证明因果关系吗? |
不能直接证明因果关系,更多是形成机制假设和筛选后续验证对象。 |
|
什么时候适合做? |
适合肠道菌群、疾病模型、药物干预、营养、环境暴露和微生态调控研究。 |
它是什么?
16S rDNA 测序通过扩增并测定细菌 16S rRNA 基因的可变区,推断样品中的微生物分类组成和相对丰度。非靶向代谢组学则使用 LC-MS、GC-MS 等平台尽可能多地采集小分子代谢物信号,通过统计分析筛选差异代谢物并进行通路解释。
联合分析的核心,是把两类数据按同一批样品或同一实验分组进行整合:先分别得到差异菌群和差异代谢物,再通过相关性分析、多变量模型、通路注释和网络图,寻找“某些菌群变化是否伴随某些代谢物变化”的证据链。
相关服务
主要优势
1、从“组成变化”走向“功能解释”
单独做 16S 分析时,研究者可以看到哪些菌增加或减少,但很难判断这些变化可能影响哪些代谢过程。加入非靶向代谢组学后,差异菌群可以与胆汁酸、短链脂肪酸、氨基酸、脂质或其他小分子变化联系起来,使结果更接近机制解释。
2、适合复杂表型的多层证据整合
疾病模型、药物干预、饮食处理和环境暴露往往同时影响微生态和宿主代谢。联合分析可以把菌群、代谢物、分组表型和通路放在一个框架中比较,帮助筛选更有价值的候选菌、候选代谢物和候选通路。
3、有利于后续验证设计
联合分析的结果常用于缩小验证范围。例如,研究者可以优先验证与关键表型相关、在多个统计方法中稳定出现、且具有生物学合理性的菌-代谢物组合,而不是盲目验证所有差异结果。
主要局限
16S 与非靶向代谢组学如何整合?
1、先分别保证单组学结果可靠
整合分析不能弥补单组学数据质量问题。16S 侧需要关注测序深度、有效序列数、稀释曲线、物种注释和样本离群;代谢组侧需要检查 QC 样本聚集、峰面积稳定性、缺失值比例、内标响应和代谢物注释等级。只有两部分结果可靠,后续相关网络才有解释价值。
2、再建立菌群与代谢物的关联关系
常见方法包括 Spearman 或 Pearson 相关分析、Mantel test、RDA/CCA、Procrustes 分析、共现网络、随机森林筛选和多组学可视化。实际选择取决于样本量、分组设计、变量数量和研究目标。对于多数科研项目,相关性网络应配合差异分析和通路背景一起解释,而不是只看相关系数大小。
3、最后回到生物学问题筛选候选机制
一个有价值的联合分析结论通常不是“某菌与某代谢物显著相关”,而是“某处理改变了特定菌群,同时影响某条代谢通路中的关键代谢物,并且这些变化与表型方向一致”。这类结果更适合作为机制假设进入靶向代谢、qPCR、菌株干预或动物实验验证。
方法选择:单做 16S、单做代谢组,还是做联合分析?
|
研究目标 |
更适合的方法 |
选择理由 |
|---|---|---|
|
只想了解菌群组成是否改变 |
16S/18S/ITS 扩增子测序 |
成本较低,适合菌群结构、差异菌和多样性分析。 |
|
只关心代谢物差异和代谢通路 |
非靶向代谢组学 |
能直接观察小分子变化,适合标志物筛选和通路探索。 |
|
想解释菌群变化如何影响代谢表型 |
16S + 非靶向代谢组联合分析 |
可以连接差异菌群、差异代谢物和表型结果。 |
|
已有明确代谢物目标 |
16S + 靶向代谢组或验证实验 |
靶向方法定量更稳定,更适合验证关键代谢物。 |
|
需要更接近功能层面的微生物解释 |
宏基因组 + 代谢组 |
比 16S 更能推断功能基因和代谢潜力,但成本和分析复杂度更高。 |

FAQ
1、16S 和非靶向代谢组学必须使用同一批样本吗?
最好使用同一批生物样本,或至少来自严格匹配的同一实验个体和时间点。联合分析依赖样本之间的一一对应关系,如果两类数据来自不同批次或不同个体,相关性解释会明显变弱。
2、联合分析需要多少样本量?
没有适用于所有项目的固定数字。一般来说,每组样本过少会让差异分析和相关分析都不稳定。实际设计应结合分组数量、预期效应大小、样本异质性、预算和后续验证计划评估。
3、相关性网络里连线越多越好吗?
不是。连线过多可能来自阈值过宽、样本量不足或多重检验控制不充分。更有价值的是与研究表型相关、统计稳定、方向合理且能被已有代谢通路或文献背景支持的核心连接。
4、非靶向代谢组学能直接确认所有代谢物吗?
不能。非靶向分析可以尽可能广泛地发现代谢物特征,但注释可信度存在等级差异。对于最终要作为标志物或机制证据的代谢物,建议进一步使用标准品、二级谱图匹配或靶向定量方法确认。
5、16S 与代谢组联合分析能证明菌群导致代谢物变化吗?
通常不能直接证明。它可以提供强机制线索,但因果关系还需要干预实验、菌株补充或去除、靶向代谢验证、同位素示踪、动物模型或体外培养体系进一步验证。
结论
16S 和非靶向代谢组学联合分析的价值,不只是把两份结果放在一起展示,而是用菌群结构、代谢物变化和实验表型共同构建机制线索。对于微生态、疾病模型、药物干预和营养研究,它能帮助研究者从“有哪些差异”进一步走向“这些差异可能如何参与生物学过程”。但联合分析仍然以数据质量、合理设计和后续验证为前提,相关性结果应作为机制假设,而不是直接等同于因果结论。
How to order?
